Research
Computational approaches to study cancer evolution.
Current & Recent Works
Epigenetic determinants of cancer cell fate
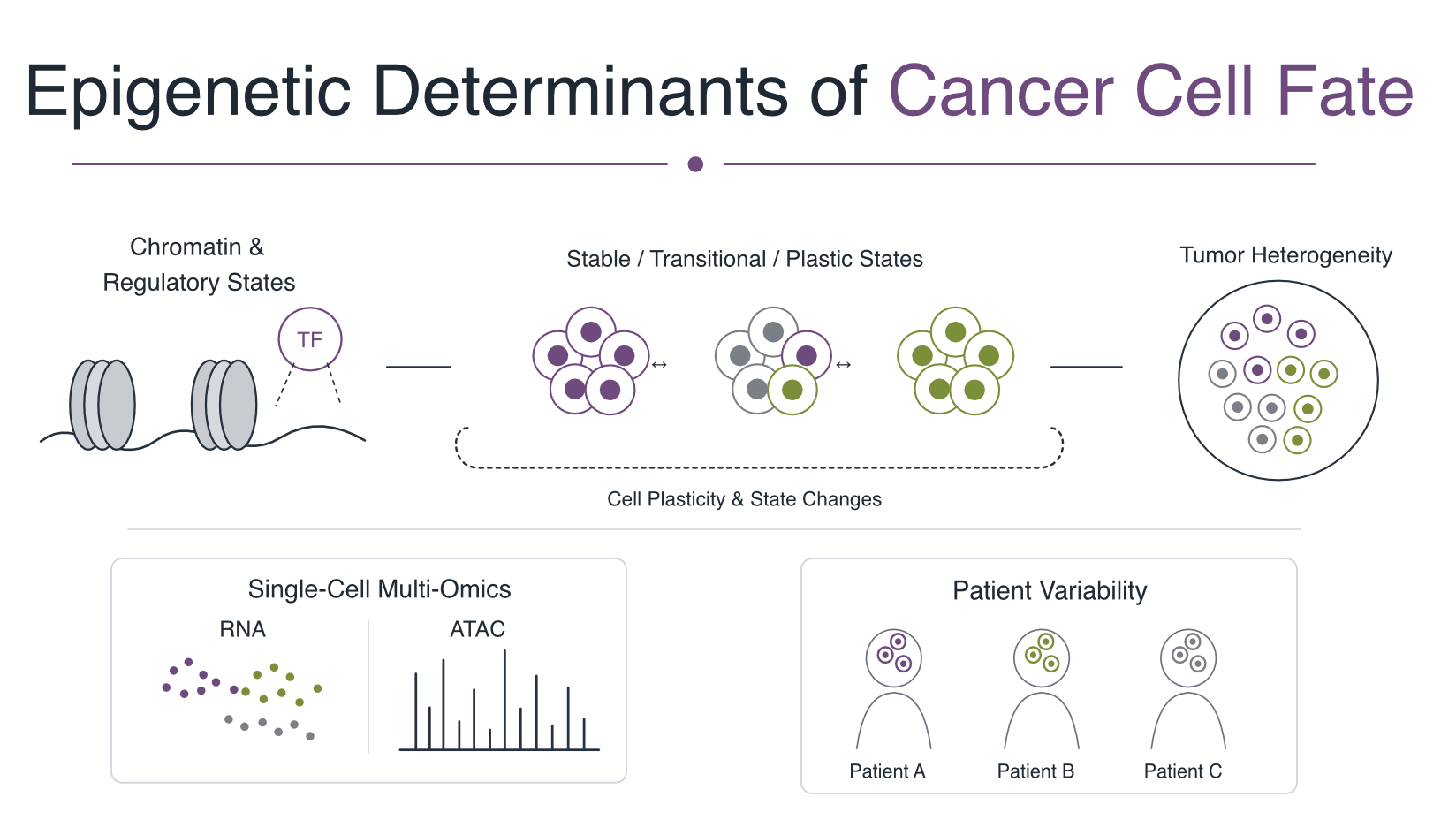
This is current, ongoing work in the Lupien Lab at Princess Margaret Cancer Centre, UHN, focused on triple-negative breast cancer (TNBC) using single-cell RNA and ATAC (multiome) data. Central questions include:
- How do chromatin and regulatory states define cancer-cell identity?
- Which cell states are stable, transitional, or plastic?
- How does inter-patient variation shape malignant cell states? Can we quanitfy such hetrogenity?
- How can single-cell multi-omic measurements reveal regulatory heterogeneity within and across tumors?
Cancer evolution and genomic dependencies

Genomic dependencies in cancer
Cancer evolution is driven by the emergence and selection of genomic alterations. As tumors grow, cancer cells accumulate genetic changes, some of which modify cellular behavior and confer a selective advantage. Determining which alterations are selected and the biological contexts in which they become advantageous is a central question in cancer genomics. My PhD research at the University of Lausanne, under the supervision of Prof. Giovanni Ciriello (lab)), focused on identifying and interpreting evolutionary dependencies through patterns of co-mutation analysis. This work built on the frameworks introduced by (Mina et al., 2020) and (Mina et al., 2022). I also developed SelectSim,a computational package for analyzing evolutionary dependencies in cancer genomic data, as part of the recently accepted manuscript, “Evolving patterns of co-mutations from tumor initiation to metastatic progression.”.
Earlier works
Before working on cancer evolution and epigenomics, I worked on several other computational biology problems:
Integrative analysis of circulating tumor cells (CTCs)
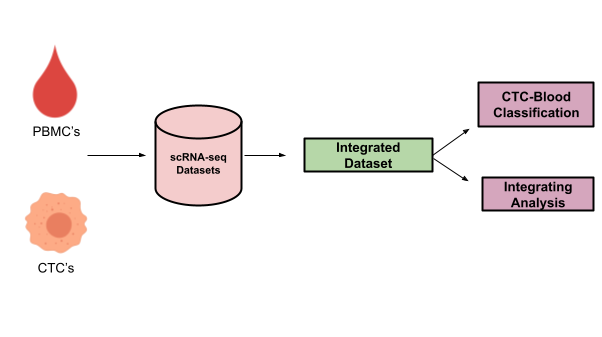
Cancer cells that detach from solid tumors and migrate through the bloodstream to colonize distant organs are called circulating tumor cells (CTCs). Under Prof. Debarka Sengupta (lab), I worked on an integrative analysis of publicly available single-cell CTC expression profiles, showing that CTCs across cancers lie on a near-continuous epithelial-to-mesenchymal (EMT) transition, and highlighting an inverse expression pattern between PD-L1 and MHC relevant to cancer immunotherapy. Published in the Journal of Clinical Medicine.
CellAtlasSearch: GPU-based search of single-cell expression profiles
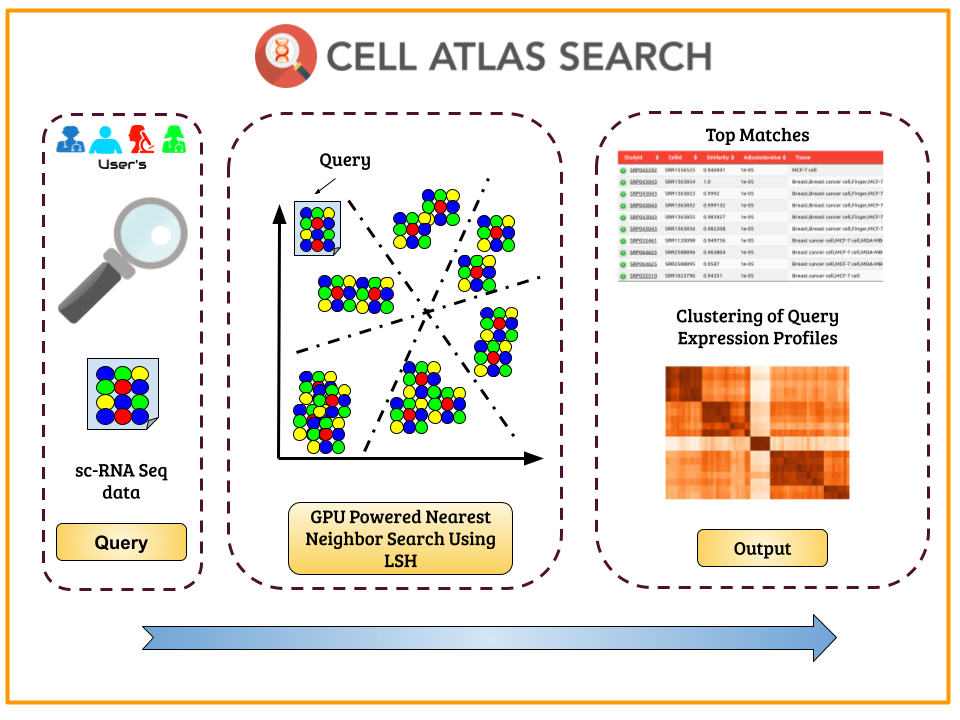
To address the need for scalable search over the growing volume of single-cell expression data, we developed CellAtlasSearch, a search architecture using GPU-accelerated Locality-Sensitive Hashing (LSH) for fast, lightweight queries over large reference expression collections (over 300,000 bulk and single-cell profiles). Co-first-authored under Prof. Debarka Sengupta (lab); published in Nucleic Acids Research.
Structural analysis of lipidated proteins
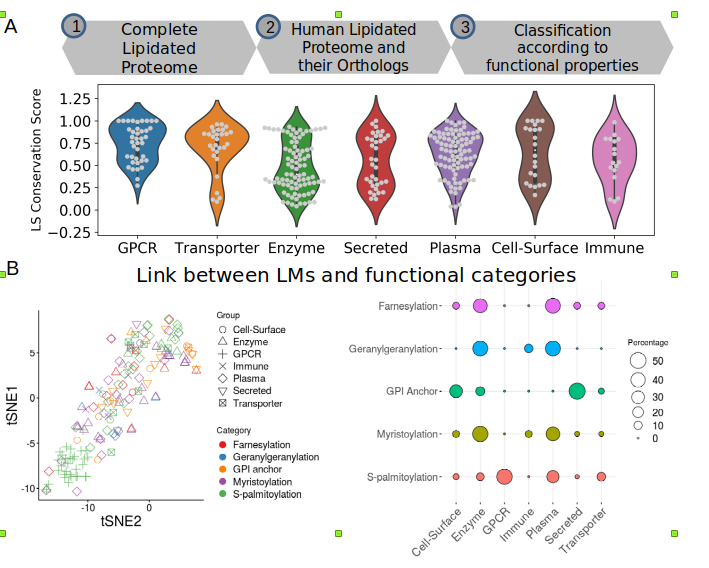
For my master’s thesis, under Prof. Lipi Thukral (lab) and Prof. Angshul Majumdar (lab), I worked on a proteome-scale analysis of lipidated proteins (a class of post-translational modification), examining how lipid-modification sites relate to protein function and location, and built a neural-network classifier to predict lipidation sites.
FlavorDB: a database of flavor molecules
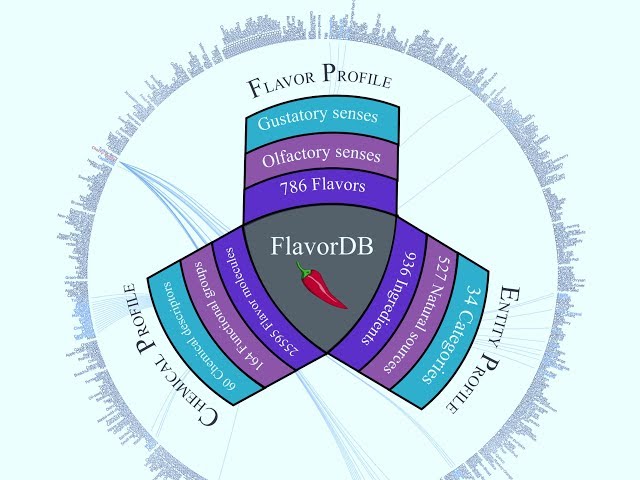
During my master’s training, I collaborated with Prof. Ganesh Bagler (lab) on foundational work in computational gastronomy. I helped build a comprehensive database of flavor molecules and their associated physicochemical properties, resulting in a co-first-author publication in Nucleic Acids Research.